What is Congenital Disorders of Glycosylation
Congenital Disorders of Glycosylation (CDG) is a rare genetic disease that affect the addition of glycans, the sugar building blocks, to proteins in cells throughout the body. This process is crucial for the proper functioning of various systems in the body. There are more than 170 types of CDG and the defects of CDG are classified into four categories: N-linked glycosylation, O-linked glycosylation, combined N- and O-linked glycosylation, and lipid and glycosylphosphatidylinositol (GPI) anchor biosynthesis.[1] However, Many categories have received so little attention that the classification of their defects remains unclear.
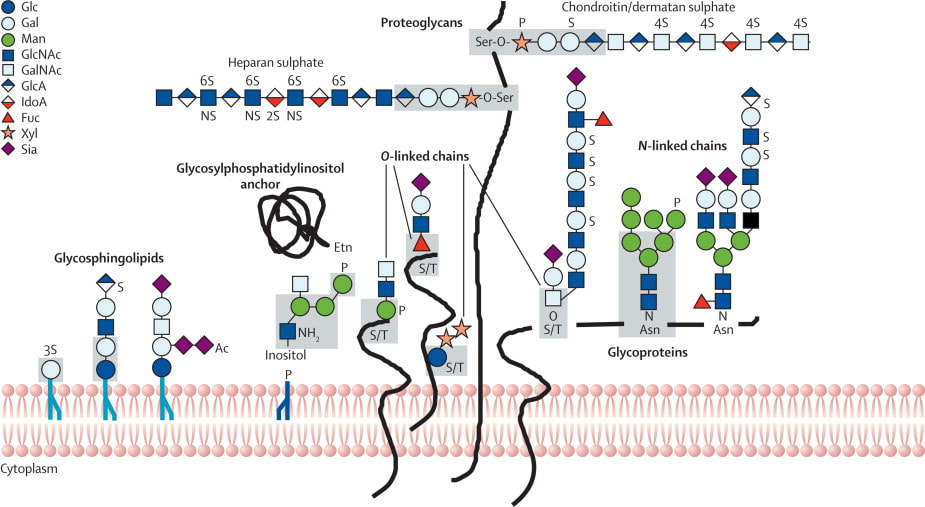
Fig 1: Different types of glycosylation
Symptoms of Congenital Disorders of Glycosylation
CDGs are characterized by a wide range of symptoms and the severity can vary greatly depending on the specific type of CDG. While many types of CDG involve central nervous system disorders, including developmental delays and brain and spine abnormalities, the condition can also affect other bodily systems. Symptoms can include but are not limited to growth failure, hormonal imbalances, muscle and liver problems, heart defects, and issues with the kidneys and bones. Gastrointestinal complications, particularly those leading to poor feeding and excessive protein loss, are also common and can contribute to developmental disabilities. [2,3]
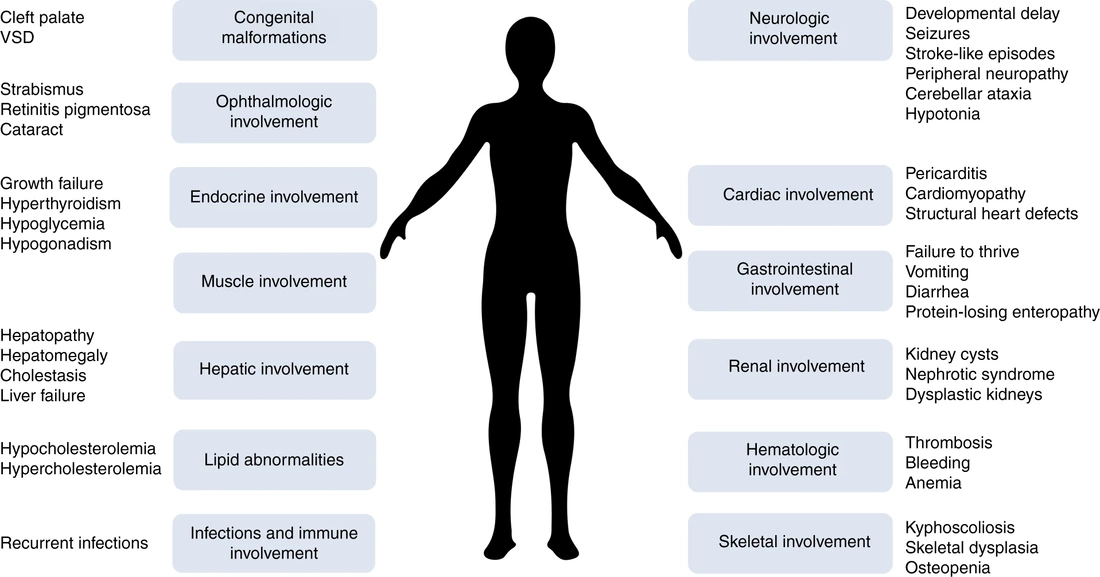
Fig 2: Symptoms of congenital disorder of glycosylation
Diagnosis and Treatment
|
|
CDG is difficult to diagnose quickly. The systemic implications of CDG necessitate a comprehensive, multidisciplinary approach for effective diagnosis and management. In addition to a thorough review of family medical histories, various physical examinations are also conducted. Traditional and common diagnostic methods include blood tests, transferrin tests—which evaluate the glycosylation status of transferrin, a key protein in iron transport—and genetic testing. For genetic tests, whole exome sequencing is popular right now as it can rapidly identify the specific type of CDG present, providing crucial information for timely treatment.[4]
Since there is no cure for CDG currently, the treatment of CDG patients mainly focus on improving the life quality. The common treatments may include specialized feeding and development therapies, seizure management via medication or surgery. Depends on the symptoms, treatments for blood clotting, vision correction, heart condition monitoring, hormonal treatments, liver function interventions and specific sugar therapies may be applied for select CDG types. For some of the SLC35A2 - CDG patients, oral D-galactose supplement is one of the potential treatment.[5] |
What is SLC35A2
The SLC35A2 (Solute carrier family 35 member A2) gene encodes the UDP-galactose transporter (UGT), which plays a critical role in glycosylation, the process of attaching sugar chains to proteins or lipids. UGT enzymes specifically transfer galactose to growing oligosaccharides at a precise step in the formation of these sugar chains. Once the appropriate number of sugar molecules are assembled, the complete oligosaccharide is attached to a protein or lipid, rendering it fully functional within the cell. Two versions of the transporter, UGT1 and UGT2, are produced by the SLC35A2 gene. [6]
How SLC35A2 is related with Congenital Disorders of Glycosylation
Mutations in the SLC35A2 gene can result in a single amino acid alteration in the UGT enzyme or disrupt its normal synthesis pathway. Consequently, this leads to the production of an abnormal UGT enzyme with reduced or entirely lost enzymatic activity. The absence of a functional enzyme prevents the completion of oligosaccharides, thereby impairing the glycosylation process and affecting the proper function of glycoproteins and glycolipids within the cell. [7,8,9,10]
The relationship between SLC35A2 mutations and specific types of glycosylation disorders remains unclear. It is unknown whether SLC35A2-CDG predominantly causes defects in N-linked, O-linked, or both N- and O-linked glycosylation pathways. Additionally, the precise impact of these mutations on various bodily systems has yet to be fully understood. This uncertainty underscores the necessity for further research to elucidate the complexities of SLC35A2-CDG and its systemic effects.[11]
Inheritance of SLC35A2 - CDG
Inheritance of SLC35A2 - CDG
|
The mutations in SLC35A2 gene are inherited in an X-linked dominant pattern, which means the gene responsible for the condition is located on the X chromosome.[12] In this pattern, if the father carries the affected X chromosome, all of his daughters will inherit the mutation and thus be affected, while his sons will not, as they inherit his Y chromosome. However, if the mother carries the affected X chromosome, each child, regardless of sex, has a 50% chance of inheriting the mutation. If the mother is unaffected, the risk is significantly reduced unless there is a new mutation. |
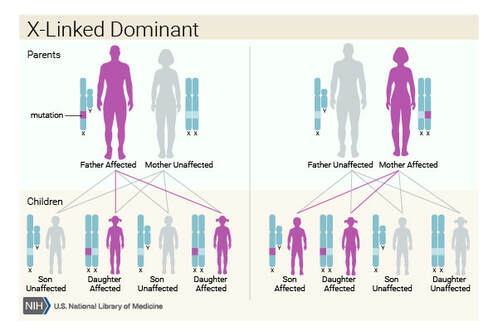
|
Organization of Congenital Disorders of Glycosylation


|

|
May 16 - World CDG Day!
Reference:
[1]Chang, I. J., He, M., & Lam, C. (2018). Congenital disorders of glycosylation. Annals of Translational Medicine, 6(24), 477. https://doi.org/10.21037/atm.2018.10.45
[2]National Organization for Rare Disorders. (2023b, January 12). Congenital Disorders of Glycosylation - Symptoms, causes, treatment | NORD. https://rarediseases.org/rare-diseases/congenital-disorders-of-glycosylation/
[3]Verheijen, J., Tahata, S., Kozicz, T., Witters, P., & Morava, É. (2020). Therapeutic approaches in Congenital Disorders of Glycosylation (CDG) involving N-linked glycosylation: an update. Genetics in Medicine, 22(2), 268–279. https://doi.org/10.1038/s41436-019-0647-2
[4]Home | CDG Hub. (n.d.). https://www.cdghub.com/
[5]Philadelphia, C. H. O. (n.d.-b). Congenital Disorders of glycosylation (CDG). Children’s Hospital of Philadelphia. https://www.chop.edu/conditions-diseases/congenital-disorders-glycosylation-cdg
[6]SLC35A2 gene: MedlinePlus Genetics. (n.d.-b). https://medlineplus.gov/genetics/gene/slc35a2/
[7]Dörre, K., Olczak, M., Wada, Y., Sosicka, P., Grüneberg, M., Reunert, J., Kurlemann, G., Fiedler, B., Biskup, S., Hörtnagel, K., Rust, S., & Marquardt, T. (2015). A new case of UDP‐galactose transporter deficiency (SLC35A2‐CDG): molecular basis, clinical phenotype, and therapeutic approach. Journal of Inherited Metabolic Disease, 38(5), 931–940. https://doi.org/10.1007/s10545-015-9828-6
[8]Quelhas, D., Correia, J., Jaeken, J., Azevedo, L., Lopes‐Marques, M., Bandeira, A., Keldermans, L., Matthijs, G., Sturiale, L., & Martins, E. (2021). SLC35A2-CDG: Novel variant and review. Molecular Genetics and Metabolism Reports, 26, 100717. https://doi.org/10.1016/j.ymgmr.2021.100717
[9]Kodera, H., Nakamura, K., Osaka, H., Maegaki, Y., Haginoya, K., Mizumoto, S., Kato, M., Okamoto, N., Iai, M., Kondo, Y., Nishiyama, K., Tsurusaki, Y., Nakashima, M., Miyake, N., Hayasaka, K., Sugahara, K., Yuasa, I., Wada, Y., Matsumoto, N., & Saitsu, H. (2013). De novo mutations INSLC35A2Encoding a UDP-Galactose transporter cause Early-Onset epileptic encephalopathy. Human Mutation, 34(12), 1708–1714. https://doi.org/10.1002/humu.22446
[10]Ng, B. G., Buckingham, K. J., Raymond, K., Kircher, M., Turner, E. H., He, M., Smith, J. D., Eroshkin, A., Szybowska, M., Losfeld, M. E., Chong, J. X., Kozenko, M., Li, C., Patterson, M. C., Gilbert, R. D., Nickerson, D. A., Shendure, J., Bamshad, M. J., & Freeze, H. H. (2013). Mosaicism of the UDP-galactose transporter SLC35A2 causes a congenital disorder of glycosylation. The American Journal of Human Genetics, 92(4), 632–636. https://doi.org/10.1016/j.ajhg.2013.03.012
[11]Reily, C., Stewart, T. J., Renfrow, M. B., & Novak, J. (2019). Glycosylation in health and disease. Nature Reviews Nephrology, 15(6), 346–366. https://doi.org/10.1038/s41581-019-0129-4
[12] SLC35A2-congenital disorder of glycosylation: Medlineplus genetic. MedlinePlus.https://medlineplus.gov/genetics/condition/slc35a2-congenital-disorder-of-glycosylation/
[1]Chang, I. J., He, M., & Lam, C. (2018). Congenital disorders of glycosylation. Annals of Translational Medicine, 6(24), 477. https://doi.org/10.21037/atm.2018.10.45
[2]National Organization for Rare Disorders. (2023b, January 12). Congenital Disorders of Glycosylation - Symptoms, causes, treatment | NORD. https://rarediseases.org/rare-diseases/congenital-disorders-of-glycosylation/
[3]Verheijen, J., Tahata, S., Kozicz, T., Witters, P., & Morava, É. (2020). Therapeutic approaches in Congenital Disorders of Glycosylation (CDG) involving N-linked glycosylation: an update. Genetics in Medicine, 22(2), 268–279. https://doi.org/10.1038/s41436-019-0647-2
[4]Home | CDG Hub. (n.d.). https://www.cdghub.com/
[5]Philadelphia, C. H. O. (n.d.-b). Congenital Disorders of glycosylation (CDG). Children’s Hospital of Philadelphia. https://www.chop.edu/conditions-diseases/congenital-disorders-glycosylation-cdg
[6]SLC35A2 gene: MedlinePlus Genetics. (n.d.-b). https://medlineplus.gov/genetics/gene/slc35a2/
[7]Dörre, K., Olczak, M., Wada, Y., Sosicka, P., Grüneberg, M., Reunert, J., Kurlemann, G., Fiedler, B., Biskup, S., Hörtnagel, K., Rust, S., & Marquardt, T. (2015). A new case of UDP‐galactose transporter deficiency (SLC35A2‐CDG): molecular basis, clinical phenotype, and therapeutic approach. Journal of Inherited Metabolic Disease, 38(5), 931–940. https://doi.org/10.1007/s10545-015-9828-6
[8]Quelhas, D., Correia, J., Jaeken, J., Azevedo, L., Lopes‐Marques, M., Bandeira, A., Keldermans, L., Matthijs, G., Sturiale, L., & Martins, E. (2021). SLC35A2-CDG: Novel variant and review. Molecular Genetics and Metabolism Reports, 26, 100717. https://doi.org/10.1016/j.ymgmr.2021.100717
[9]Kodera, H., Nakamura, K., Osaka, H., Maegaki, Y., Haginoya, K., Mizumoto, S., Kato, M., Okamoto, N., Iai, M., Kondo, Y., Nishiyama, K., Tsurusaki, Y., Nakashima, M., Miyake, N., Hayasaka, K., Sugahara, K., Yuasa, I., Wada, Y., Matsumoto, N., & Saitsu, H. (2013). De novo mutations INSLC35A2Encoding a UDP-Galactose transporter cause Early-Onset epileptic encephalopathy. Human Mutation, 34(12), 1708–1714. https://doi.org/10.1002/humu.22446
[10]Ng, B. G., Buckingham, K. J., Raymond, K., Kircher, M., Turner, E. H., He, M., Smith, J. D., Eroshkin, A., Szybowska, M., Losfeld, M. E., Chong, J. X., Kozenko, M., Li, C., Patterson, M. C., Gilbert, R. D., Nickerson, D. A., Shendure, J., Bamshad, M. J., & Freeze, H. H. (2013). Mosaicism of the UDP-galactose transporter SLC35A2 causes a congenital disorder of glycosylation. The American Journal of Human Genetics, 92(4), 632–636. https://doi.org/10.1016/j.ajhg.2013.03.012
[11]Reily, C., Stewart, T. J., Renfrow, M. B., & Novak, J. (2019). Glycosylation in health and disease. Nature Reviews Nephrology, 15(6), 346–366. https://doi.org/10.1038/s41581-019-0129-4
[12] SLC35A2-congenital disorder of glycosylation: Medlineplus genetic. MedlinePlus.https://medlineplus.gov/genetics/condition/slc35a2-congenital-disorder-of-glycosylation/
